Publications:
- Shi G, Kang X, Dong F, Liu Y, Zhu N, Hu Y, Xu H, Lao X, Zheng H. DRAMP 3.0: an enhanced comprehensive data repository of antimicrobial peptides. Nucleic Acids Res. 2022 Jan 7;50(D1):D488-D496. PMID: 34390348
- Kang X.; Dong F.; Shi C.; Zheng H. et al. DRAMP 2.0, an updated data repository of antimicrobial peptides. Scientific Data. 2019; 6(1): 148. PMID:31409791
- Liu S.; Bao J.; Lao X.; Zheng H. Novel 3D Structure Based Model for Activity Prediction and Design of Antimicrobial Peptides. Sci Rep. 2018 Jul 25;8(1):11189. PMID:30046138
- Liu S.; Fan L.; Sun J.; Lao X.; Zheng H. Computational resources and tools for antimicrobial peptides. J Pept Sci. 2017 Jan;23(1):4-12. PMID:27966278
- Fan L.; Sun J.; Zhou M.; Zhou J.; Lao X.; Zheng H.; Xu H. DRAMP: a comprehensive data repository of antimicrobial peptides. Sci Rep. 2016 Apr 14;6:24482. PMID: 27075512
Contents
Dataset
Search help
Tools help
General Data
General dataset is the most important part of DRAMP because each entry has detailed annotations and serious descriptions. We collect information of General AMPs from journal papers, which means reliability and traceability to some extent. General dataset can be further divided into two subsets: Natural AMPs and Synthetic AMPs. Natural AMPs are originally isolated or extraced from nature and synthetic AMPs are manually synthesized initially (including AMPs derived from natural AMPs). We provide Uniprot ID and PDB ID for natural AMPs (if they have) in purpose of presenting more data sources.
Fileds
General Information
- DRAMP ID >>> The field provides the unique accessing number linking to the corresponding DRAMP entry (Letters "DRAMP" followed by a 5-digit number). DRAMP IDs are supposed to be allocated in a continuous order from small to large, but some DRAMP IDs are empty thanks to removal of redundant entries or our negligence. We don't plan to allocate these medial empty DRAMP ID to new entries so as not to confuse the addition order of entries. e.g. DRAMP00001.
- Peptide Name >>> The field presents the name of each peptide in DRAMP. The names of natural AMPs are given according to their entry name in Uniprot and sysnthetic AMPs are named on the basis of literature content. e.g. Esculentin-1Vb or esc1Vb.
- Source >>> The field shows where the peptide came from initially. For natural AMPs, Source means the organism where the peptides were extracted or isolated. The sources of synthetic AMPs are the same: synthetic construct. e.g.Rana palustris or Rana palustris (Pickerel frog) or just Frog.
- Family >>> The field provides information on sequence similarities with other peptides or proteins. The field is not applicable to synthetic AMPs. e.g.Belongs to the gastrin/cholecystokinin family. Magainin subfamily.
- Gene >>> The field indicates the name of the gene that code for the peptide sequence described in the entry. Similar to the Family field, Gene is not applicable to synthetic AMPs. e.g.magainins.
- Sequence >>> The field displays the peptide sequence which is represented by single letter codes. It only accepts the mature part of peptide and terminal modifications are not included in the field. There is no space in the sequence for the purpose of retrival convenience. e.g.GIGKFLHSAKKFGKAFVGEIMNS.
- Sequence Length >>> The field tells the number of resiudes in the peptide sequence. e.g.15.
- Uniprot Entry >>> The filed provides the accessing link(s) directing to external Uniprot entry(or entries). Different Uniprot entry in the sanme DRAMP entry is separated by ','. e.g.P84930 or P84930,P84278.
- Protein Existence >>> The field indicates the type of evidence that supports the existence of the peptide. It is only applicable to the peptide which has an entry in Uniprot. e.g.Experimental evidence at protein level or Experimental evidence at transcript level or peptide inferred from homology or peptide predicted or peptide uncertain.
Activity Information
- Biological Activity >>> The field summarizes the known function(s) of the peptide entry according to literature. You can see the activity relationship map in "Read more" page. e.g. Antimicrobial, Antibacterial, Anti-cancer, Anti-Gram+
- Target Organism >>> The field displays biological activity information against microbials or cancer cells, which is consist of reference, target organism(s) and corresponding minimum inhibition concentration. e.g. [Ref.XXXXXXX]Gram-negative bacteria: Escherichia coli (MIC = 5 μg/mL).
- Hemolytic Activity >>> The field displays hemolytic activity information against red blood cells (RBCs), which is consist of hemolysis and target RBCs. e.g.[Ref.22029824] It shows no hemolytic activity against human red blood cells up to the concentration of 50 μg/mL.
- Cytotoxicity >>> The field displays cytotoxicity information against host cells except RBCs, which is consist of cytotoxicity and target host cells. e.g.[Ref.31133658] The cell viability of MDMs induced by Pep-H is 94.2%, 91.5%, 90.4%, 88.0% and 84.1% at peptide concentrations of 1, 5, 25, 50, 100 μg/mL.
- Binding Target >>> The field displays the action site of AMPs against target organism cells. e.g.Cell membrane.
Structure Information
- Linear/Cyclic >>> The field shows whether the peptide is linear or cyclic. e.g.Linear.
- N-termincal Modification >>> The field shows whether and what N-terminal modifications the peptide have according to the reference. e.g.Acetylation
- C-termincal Modification >>> The field shows whether and what C-terminal modifications the peptide have according to the reference. e.g.Amidation
- Other Modifications and Unusual Amino Acids >>> The field presents all bonds and special amino acids (out of 20 common amino acids). e.g.Disulfide bond between Cys27 and Cys33. or The 'J' in the sequence is the 2-naphthyl alanine (2-Nal) residue.
- Stereochemistry >>> The field shows whether the peptide is consist of some or all D-amino acids. e.g.L or D or Mixed (D-Leu9, D-Lys10)
- Structure >>> The field shows the overall secondary structure of the peptide. e.g.α-helix or Random coil
- Structure Description >>> The field presents the original text about secondary structure of the peptide. It often means a structure comparison with other derived AMPs. e.g.The CD spectra for all the peptides in 10 mM sodium phosphate buffer (pH 7.2) displayed a negative band at approximately 200 nm, indicating that the structure is random. In presence of 0.1% LPS, all the peptides showed characteristic α-helical CD spectra with two dichroic minimal values at 208 and 222 nm and a positive band near 192 nm .
- PDB ID >>> The field provides accessing link(s) directing to the correspong PDB entry. Users can also see the PDB figure directly in the page. We also provides predicted structure of the peptide with the help of MOE and Amber. e.g. 1OF9 resolved by NMR.
- Helical wheel diagram
Physicochemical Information
The section contains formula, mass, pI, Net charge and other information. We use Expasy and Scientific Database Maker to get such information. Notes: Terminal modifications are not considered in the calculation of formula and mass.
Comment Information
- Function >>> The field presents our comments on the AMP, which may include the stability, background information and antibacterial activity comparison with other analogs
- PTM >>> This subsection of the PTM/processing section describes post-translational modifications (PTMs). This subsection complements the information provided at the sequence level or describes modifications for which position-specific data is not yet available.
Literature Information
The information of General AMPs come from all kinds of literature and the section provides the way to find the full text.
Patent Data
Patent dataset is based on a large amount of patent sequences, which account for a large proportion in DRAMP. Such patent AMPs information can show existing patented AMP sequences and help researchers avoid infringement risk. However, patent AMPs have not been peer-reviewed, which means many sequences referred may not be really antimicrobial.
The page of patent AMPs present patent accession number and family information. Users can browse detailed patent information in Lens.org
Clinical Data
Clinical trials dataset is an important part of our database, although the clinical data are fewer comparing to other datasets. It is difficult for us to track clinical trial information in real time and it means some data are out of date. We are looking forward to reconstruct the page of clinical data in the near future.
Specific Data
Specific dataset contains stapled AMPs and HTS AMPs. Stapled peptides are special and novel artificial modified peptides, so their information is presented in a more simplified and specific manner than general entries. ome inessential fields were removed, such as source, family, gene, UniProt entry and evidence code. Meanwhile, the original sequence and predicted structure diagram of stapled AMP are shown.
High-throughput screening and in silico methods establish an innovative approach to comparatively quickly design novel antimicrobials. To expand the retrieval scope and assist in the design of AMPs, we added thousands of candidate AMPs screened by a high-throughput platform.
Stable Data
To further promote the clinical translation of AMPs and assist researchers in designing and selecting effective AMP drugs, DRAMP 4.0 includes the collection of AMP entries with experimentally validated stability data not yet included in any existing online AMP databases. The stability data section provides three types of annotations: the type of serum or protease, stability data, and methods used for stability determination, along with links to relevant literature.
Expanded Data
We have added the Expanded AMPs dataset, which comprises experimentally verified AMPs from Chinese literature.
Quick Search
Quick search allows keywords searches to performed on all text fields:
- Identify the keywords of interest for your search.
- Enter the terms (or key concepts) in the search box.
- Click "Quick Search".
Simple Search
The simple search page allows you to search individual fields found within antimicrobial peptides (AMPs).
- Find a list of all indexed fields in the drop down menu and choose one of your interested.
- Enter the appropriate contents in the text area below.
- Click "Submit" (or click "Reset" to clear your input).
- DRAMP ID >>> Accessing numble and linking to the DRAMP entries (Letters "DR" followed by a 5-digit number).e.g. DRAMP00001
- Peptide Name >>> Name of peptides in DRAMP (full name or short name works). e.g. Esculentin-1Vb or esc1Vb
- Sequence >>> Single letter code (no space, mature peptide only).e.g. AVPAVRKTNETLD
- Source >>> Scientific name of the source organism of AMPs (species).e.g. Rana palustris or Pickerel frog or Rana palustris (Pickerel frog) or just frog
- Family >>> Defined protein family (subfamily).e.g. Plant LTP family, Alpha defense family
- Swiss-prot Entry >>> Accessing number and linking to UniProtKB/Swiss-Prot entries (If you want to search multiple entries at the same time, entries must be separated by ,).e.g. P84930 or P84930,P84278
- PDB ID >>> Accessing numble of Protein Data Bank. e.g. 1JMN or 1JMN,1OKH
- Pubmed ID >>> A unique identifier of each record in PubMed (PMID for short). e.g. 12600207 or 12600207,19111587
- Literature citation >>> Literature information about AMPs, including Reference, Author and Title.
- Patent No. >>> A unique identifier of Patented AMPs. e.g. US 8334366 (US granted patents) or EP 383808 (European granted patents)
- Clinical rials.gov Indentifier >>> A unique identification code given to each clinical study registered on ClinicalTrials.gov. (Letters "NCT" followed by an 8-digit number). e.g. NCT00000419
Advanced Search
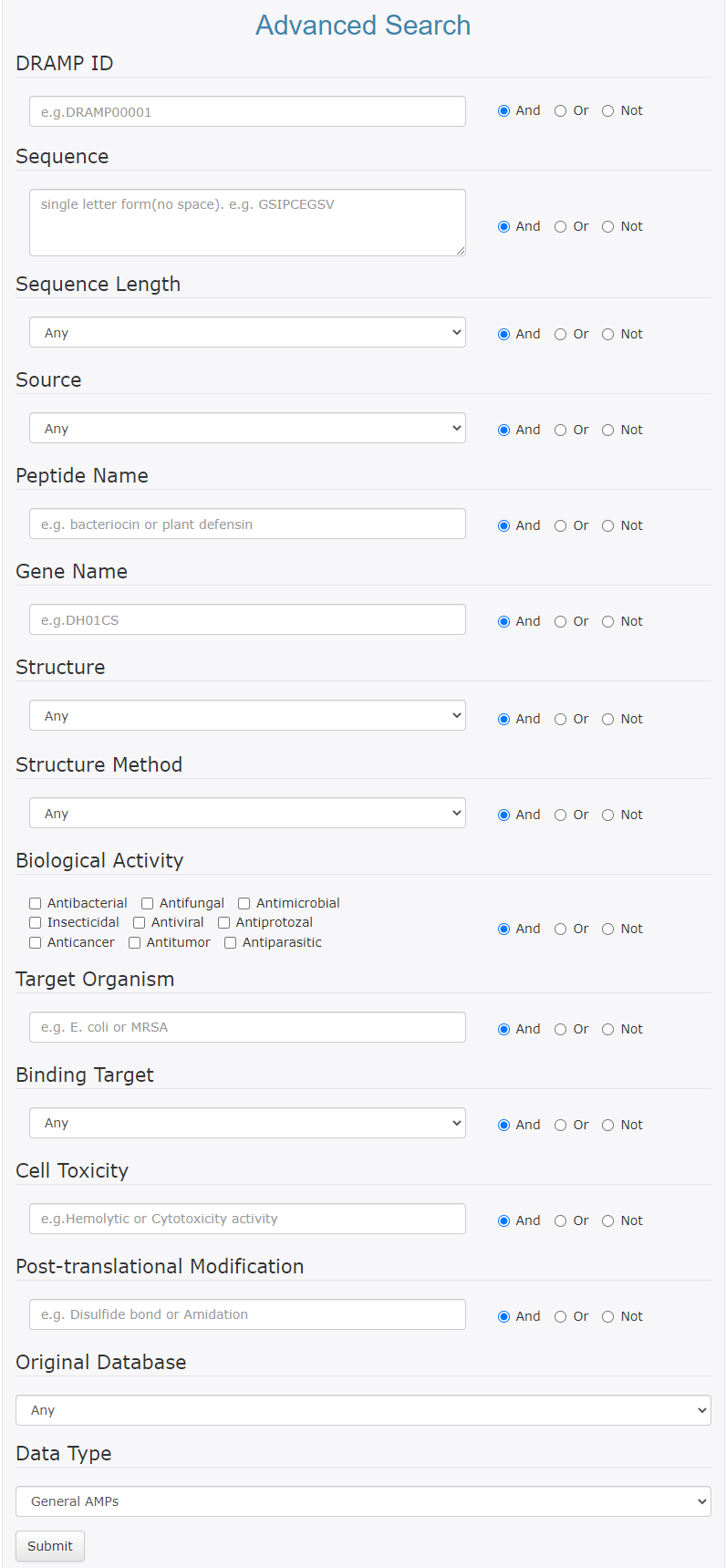
Currently we support the AND, OR and NOT search operators, which tell the DRAMP how the words in your search are related to each other.
- Use AND to find study records that contain all the words connected by AND.
- Use OR to find study records that contain either word connected by OR.
- Use NOT to find study records that do not contain the word following NOT.
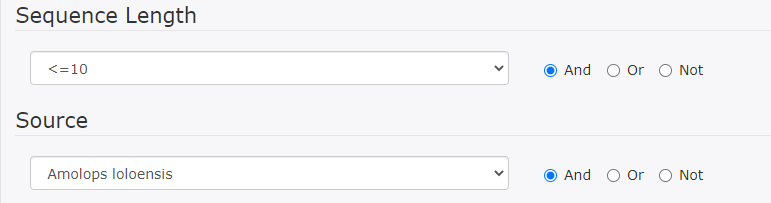
This search finds DRAMP entries that contain information on both sequence length (<=10) and source (Amolops loloensisi).
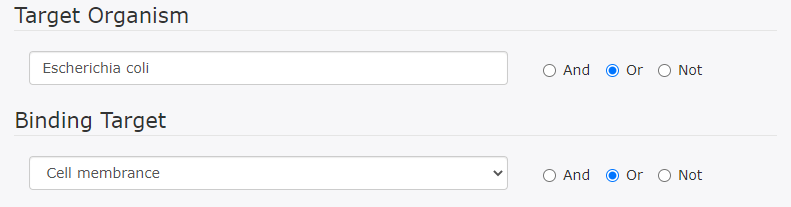
This search finds DRAMP entries containing either the word "Escherichia coli" or the word "Cell membrane".
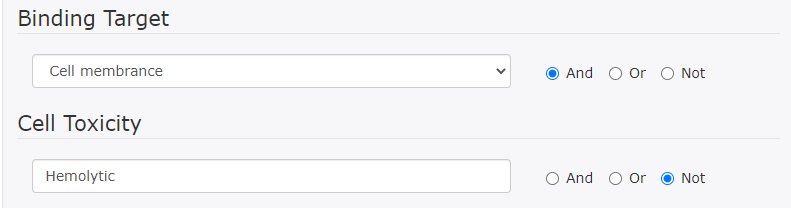
This search finds DRAMP entries containing the word "Cell membrane" but excludes entries containing the word "Hemolytic" from the search results.
Similarity Search (Local alignment)
The BLAST (Basic Local Alignment Search Tool) program uses a strategy based on matching sequence fragments by employing a powerful statistical model to find the best local alignments (For more information see http://www.ebi.ac.uk/Tools/sss/ncbiblast/).
The Smith–Waterman search is implemented using the SSEARCH program from the FASTA distribution (For more information see http://fasta.bioch.virginia.edu/). BLAST is a faster but less accurate method than SSEARCH.
Usage Introduction
Step 1 – Sequence Input
- Sequence Input Window: The query sequence can be entered directly into text area. The sequence must be FASTA format.
- Sequence File Upload: A file containing a valid sequence in FASTA format can be used as input for the sequence similarity search.
FASTA format: FASTA formatted sequence records start with a definition line, which must start with a > character. The definition line must occupy one single line and followed by sequence data.
Example:
>Antimicrobial peptide LCI
AIKLVQSPNGNFAASFVLDGTKWIFKSKYYDSSKGYWVGIYEVWDRK
Step 2 – Database
Select the protein sequence database to run the sequence similarity search against your input sequence.
Default value is: DRAMP
Step 3 – Parameters
- Matrix: This option allows you to choose the scoring matrix to be applied to the search.
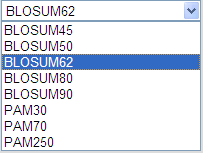
Default value is: BLOSUM62
Tip: In general, higher value BLOSUM matrices (e.g. BLOSUM90) and lower value PAM matrices (e.g. PAM30) are more stringent than low value BLOSUM or high value PAM matrices. This implies that if you want to find more distantly related homologues, you should preferentially employ a low value BLOSUM or high value PAM matrix (For more information about scoring matrices see http://en.wikipedia.org/wiki/Matrix).
Multiple sequence alignment
Clustal Omega is a new multiple sequence alignment program that uses seeded guide trees and HMM profile-profile techniques to generate alignments (For more information see http://www.ebi.ac.uk/Tools/msa/clustalo/).
MUSCLE stands for multiple sequence comparison by log-expectation. MUSCLE is claimed to achieve both better average accuracy and better speed than ClustalW2 or T-Coffee (For more information see http://www.ebi.ac.uk/Tools/msa/muscle/).
Usage Introduction
Step 1 – Sequence input
- Sequence Input Window: Three or more sequences to be aligned can be entered directly into this box. The sequences must record in FASTA format,which begins with a defined line. The defined line must start with a '>' character.
- Sequence File Upload: A file containing a valid sequence in FASTA format can be used as input for the sequence similarity search.
Step 2 – Submission
Job Title: It's possible to identify the tool result by giving it a name. This name will be associated to the results and might appear in some of the graphical representations of the results.
Protein conserved domain search (CD-search)
CD-Search is a tool for the detection of conserved domains in protein sequences. It can therefore help to elucidate the protein's function. The CD-Search service uses RPS-BLAST to compare a query protein sequence against conserved domain models that have been collected from a number of source databases.
RPS-BLAST, which stands for "Reverse Position-Specific BLAST". This is a variant of the popular PSI-BLAST program ("Position-Specific Iterated BLAST").For more information see http://www.ncbi.nlm.nih.gov/Structure/cdd/cdd_help.shtml.
Usage Introduction
Step 1 - sequence input
The query sequence can be entered directly into text area. The sequence must be in FASTA format. A file containing a valid sequence in FASTA format can also be used as input for the conserved domain search.
Step 2 - Search parameters
Database Selection
- Expect Value (E-value): E-value describes the random background noise that exists for matches between sequences. The lower the E-value, or the closer it is to "0", the higher is the "significance" of the match.
- Maximum number of hits: limits the size of the hit list produced by CD-Search.
Step 3 – Submission
For more information see: http://www.ncbi.nlm.nih.gov/Structure/cdd/cdd_help.shtml
